
 X
X Facebook
Facebook LinkedIn
LinkedIn Reddit
Reddit Bluesky
BlueskyCrystallographic insights into the hydrogen barrier mechanism of polyethylene furanoate (PEF) for high-pressure storage applications: comparison with polyamide 6 and polyethylene
- Volume
- CitationLiu Z, Guo Y, Gu B, Qiu N, Bai X, et al. Crystallographic insights into the hydrogen barrier mechanism of polyethylene furanoate (PEF) for high-pressure storage applications: comparison with polyamide 6 and polyethylene. AI Mater. 2025(2):0013, https://doi.org/10.55092/aimat20250013.
- DOI10.55092/aimat20250013
- CopyrightCopyright2025 by the authors. Published by ELSP.
- Special Issue
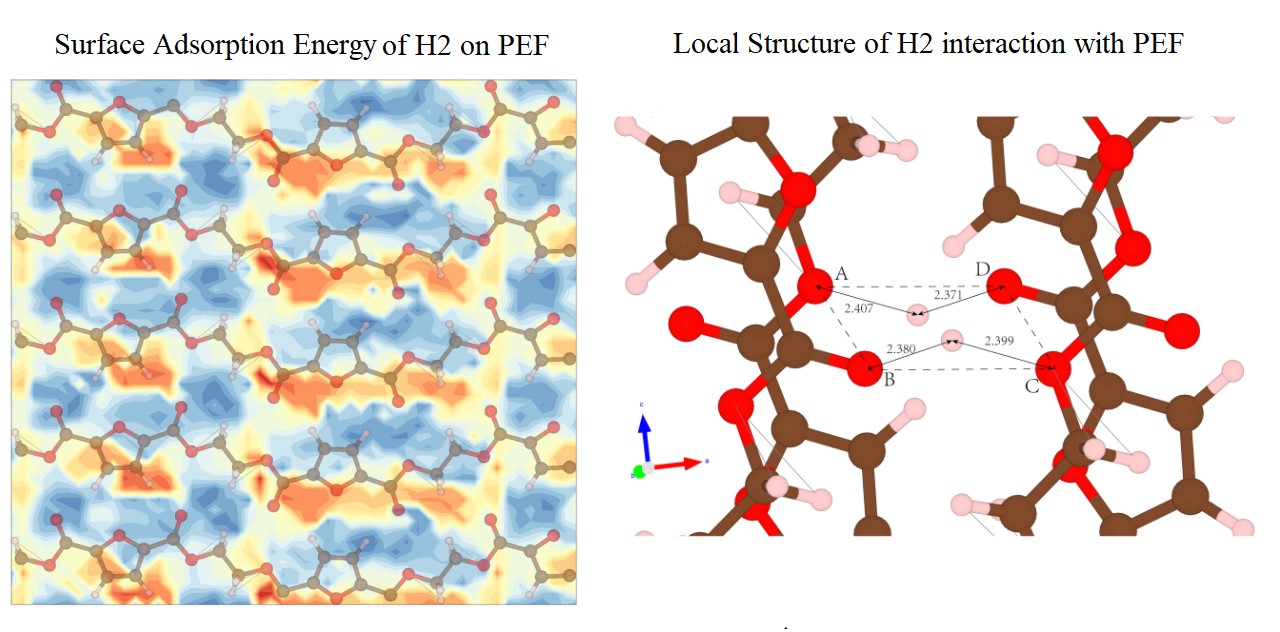
This work examines the hydrogen barrier capabilities of the crystalline α-PEF relative to α-PA6 and α-PE through MD simulations using a revised CVFF force field benchmarked against DFT calculations. Although minor deviations persist in chain-level details, our MD model successfully reproduces the key crystallographic and energetic properties of α-PEF, including lattice constants, surface energy, and NEB barriers. DFT results show that α-PEF exhibits superior polymorphic stability under pressure, with a potential stress-induced transition from α′ to α. MD simulations reveal weak hydrogen physisorption in the three materials, but PEF shows relatively stronger adsorption at low temperatures due to the oxygen-rich surface, accompanied by enhanced van der Waals and coulombic interactions with H₂. According to NEB calculations, particularly, PEF consistently possesses the highest H₂ migration barriers, that is, 0.772 eV (surface entry), 0.555 eV (escape), and 0.828 eV (bulk diffusion), exceeding those of PA6 and PE. The unique behavior of PEF stems in part from its high bulk density, but more distinctively from the quasi-coplanar molecular traps formed by intrachain oxygen atoms that impede the movement of H₂ molecules. Despite the risks associated with cryogenic adsorption, the crystalline structure of α-PEF provides the basis for its remarkable hydrogen barrier properties, making it an excellent candidate for practical high-pressure hydrogen applications.
PEF crystal; molecular simulation; hydrogen barrier; CVFF force field; gas transport
